Fabry disease is a rare, X-linked inherited disorder caused by a deficiency or absence of the lysosomal enzyme α-galactosidase A (α-Gal A). Without this enzyme, the body cannot properly break down certain fatty substances, leading to the buildup of globotriaosylceramide (Gb3) and related glycosphingolipids inside cells. Over time, this accumulation disrupts normal cell structure and function, affecting multiple organs. As the disease progresses, it can lead to serious complications such as stroke, heart failure, cardiac arrhythmias, and end-stage renal disease, ultimately reducing life expectancy in affected individuals.
This article explores its underlying causes, key symptoms, current treatment options, and emerging therapeutic approaches – including advanced organoid models – that are shaping the future of research and patient care.
In This Article

What Causes Fabry Disease?
Fabry Disease is caused by mutations in the GLA gene, which result in reduced or absent activity of the enzyme alpha-galactosidase A. Without sufficient levels of this enzyme, certain fatty substances cannot be properly broken down and gradually accumulate within cells over time.
The condition is inherited in an X-linked pattern, meaning the gene responsible is located on the X chromosome. As a result, both males and females can be affected, although males typically experience more severe symptoms due to having only one X chromosome. Fabry Disease can present in different forms, including a classic early-onset type that appears in childhood and a later-onset form that may develop in adulthood with more variable symptoms.
Symptoms of Fabry Disease
The symptoms of Fabry Disease vary widely and often begin in childhood or adolescence, although some individuals may not develop noticeable signs until adulthood. Early manifestations commonly include episodes of burning or tingling pain in the hands and feet (acroparesthesia) and reduced sweating (hypohidrosis). Many patients also develop small, dark red skin lesions called angiokeratomas, as well as gastrointestinal issues such as abdominal pain and diarrhea. Eye changes, including corneal opacity, may occur but often do not significantly affect vision.
As the disease progresses, the accumulation of fatty substances in cells can lead to more serious complications involving major organs. Kidney function may gradually decline, potentially resulting in kidney failure, while the heart can be affected by conditions such as cardiomyopathy and arrhythmias. In addition, individuals with Fabry Disease are at an increased risk of stroke or transient ischemic attacks.
Late-onset cases may only show heart or kidney symptoms in adulthood. Early recognition of these symptoms is crucial for timely diagnosis and treatment.
Because these symptoms can overlap with those of other conditions and vary greatly between patients, Fabry Disease is often underdiagnosed or misdiagnosed, especially in its early stages.
Read more:
How is Fabry Disease Diagnosed?
Diagnosing Fabry disease involves a combination of clinical evaluation, enzyme testing, and genetic analysis. In males, a simple blood test measuring alpha-galactosidase A (α-Gal A) enzyme activity is usually sufficient. Very low or absent enzyme levels strongly indicate Fabry disease. In females, enzyme levels can be normal or near-normal, so GLA gene sequencing (genetic testing) is required to identify disease-causing mutations.
Additional supportive tests may include measuring lyso-Gb3 levels in blood, a slit-lamp eye exam for corneal verticillata, or organ imaging. Early and accurate diagnosis is essential to start treatment before irreversible kidney, heart, or neurological damage occurs.
Current Treatment Options
Current treatments for Fabry Disease focus on managing symptoms and slowing disease progression. Enzyme replacement therapy (ERT) provides a synthetic form of alpha-galactosidase A to help reduce the buildup of harmful substances in cells, while pharmacological chaperone therapy works by stabilizing the patient’s own defective enzyme in certain cases. In addition, supportive care such as pain management, cardiovascular monitoring, and renal support, including dialysis or transplantation, plays an important role in improving quality of life.
Emerging Therapies and Research Advances
Gene therapy
Gene therapy is being actively explored as a potential long-term solution for Fabry Disease by introducing a functional copy of the GLA gene into patients’ cells. This approach aims to restore the body’s ability to produce the missing enzyme, potentially reducing or eliminating the need for lifelong treatments.
Substrate reduction therapy (SRT)
Substrate reduction therapy (SRT) offers another promising strategy by targeting the production of globotriaosylceramide (Gb3). Instead of replacing the enzyme, this approach reduces the amount of substrate that accumulates in cells, helping to slow disease progression and complement existing therapies.
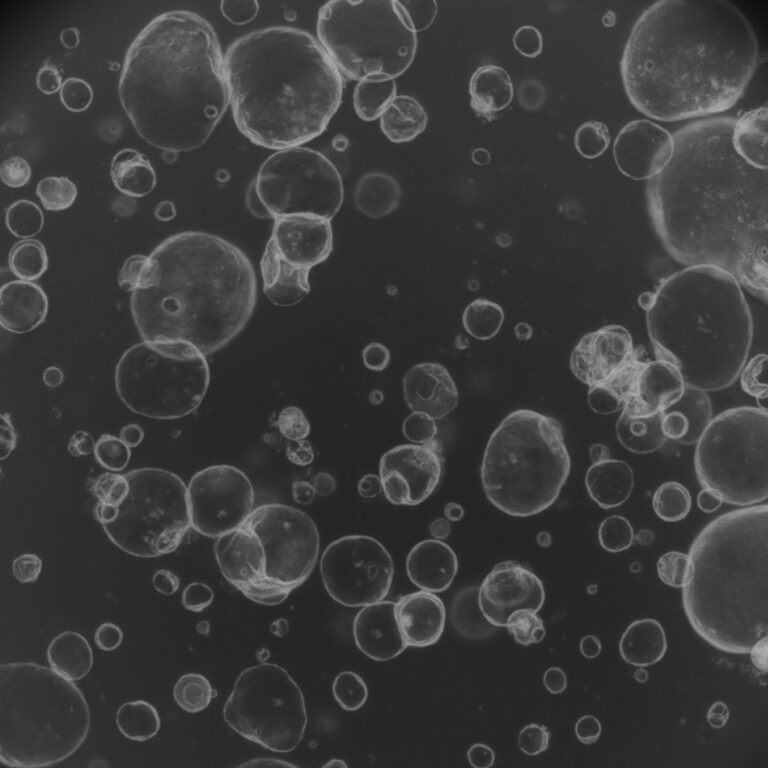
Kidney Organoid Models in Fabry Disease Research
Accurate Fabry disease modelling is crucial because traditional animal models often fail to fully replicate human renal pathology. A groundbreaking 2021 study published in Experimental & Molecular Medicine addressed this by creating human kidney organoids from CRISPR-Cas9-edited iPSCs with GLA knockout. These 3D kidney organoids faithfully reproduced key features of Fabry nephropathy, including massive Gb3 accumulation, characteristic “zebra bodies” under electron microscopy, podocyte and tubular cell deformation, oxidative stress, mitochondrial dysfunction, and increased apoptosis.
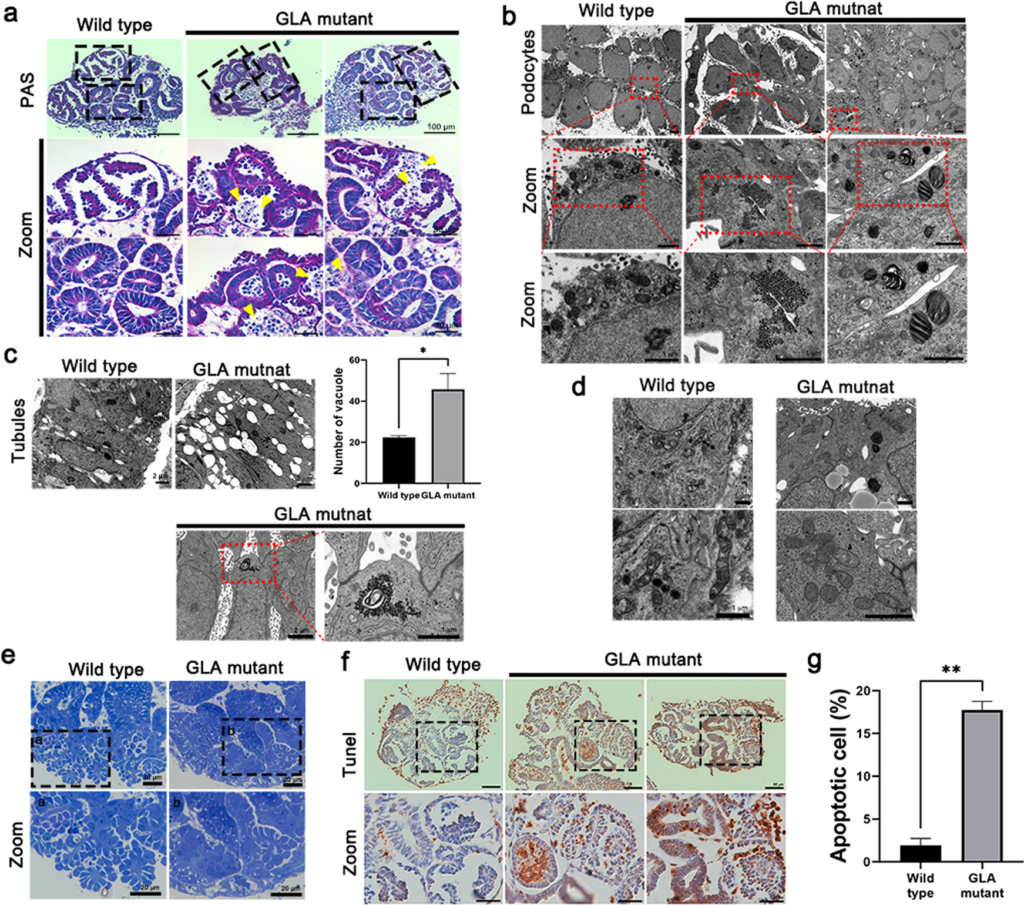
Transcriptomic analysis further revealed disrupted glutathione (GSH) metabolism as a central disease mechanism. The model also demonstrated that ERT partially cleared Gb3, while GSH supplementation offered additional protective effects by reducing oxidative stress and improving cellular markers.
Lambda Biologics Solution
Lambda has developed the kidney organoid model for Fabry’s disease using CRISPR-Cas9 gene editing. This model closely mirrors the pathological phenotype of human Fabry disease and responds to enzyme replacement therapy. Consequently, it stands as a valuable tool in the development of treatments for Fabry disease.
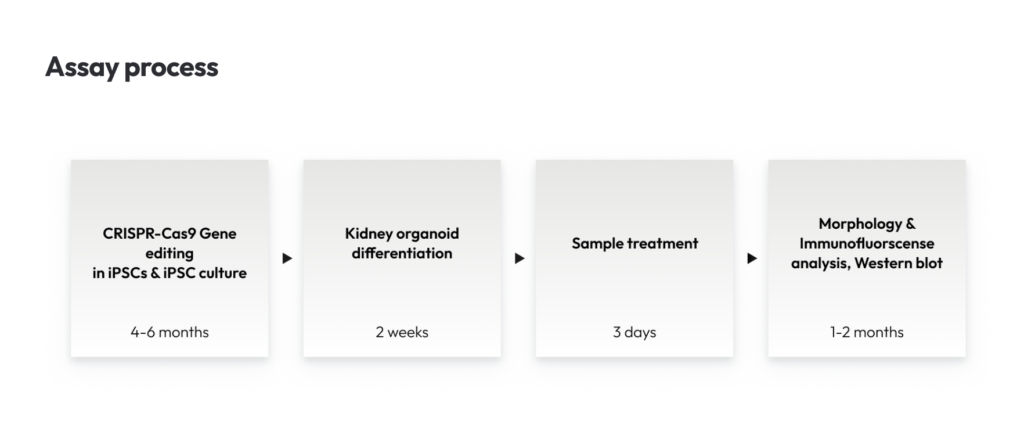
Conclusion
Early detection of Fabry Disease is critical for slowing disease progression and preventing irreversible organ damage. Because symptoms are often nonspecific and vary widely between individuals, diagnosis is frequently delayed, reducing the window for effective intervention. Identifying the disease early allows for timely initiation of treatment and better long-term outcomes. As advances in gene therapy, targeted treatments, and human-relevant research models continue to evolve, the outlook for Fabry Disease is steadily improving, offering new hope for more effective and personalized management in the future.
Research article: Human kidney organoids reveal the role of glutathione in Fabry disease
Lambda Biologics’ Organoid-based Evaluation Platforms
Gastric Cancer Organoid | Kidney Organoid | Breast Cancer Organoid | Hepatocarcinoma Cancer Organoid | Pancreatic Cancer Organoid